
Inhaled alpha-1-antitrypsin (AAT) restores lower respiratory tract protease- anti-protease homeostasis and reduces inflammation in AAT- deficient individuals: A randomised phase 2 study
Mark Brantly, James Stocks, Jorge Lascano, Tammy Flagg, Ann M. Jeffers, Shuzi Z. Owens, Torry A. Tucker, Megan Devine, Noga Alagem, Naveh Tov
Please cite this article as: Brantly M, Stocks J, Lascano J, et al. Inhaled alpha-1-antitrypsin (AAT) restores lower respiratory tract protease- anti-protease homeostasis and reduces inflammation in AAT-deficient individuals: A randomised phase 2 study. ERJ Open Res 2024; in press (https://doi.org/10.1183/23120541.00537-2024).
This manuscript has recently been accepted for publication in the ERJ Open Research. It is published here in its accepted form prior to copyediting and typesetting by our production team. After these production processes are complete and the authors have approved the resulting proofs, the article will move to the latest issue of the ERJOR online.
Copyright ©The authors 2024. This version is distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0. For commercial reproduction rights and permissions contact permissions@ersnet.org
Inhaled alpha-1-antitrypsin (AAT) restores lower respiratory tract protease- anti-protease homeostasis and reduces inflammation in AAT-deficient individuals: A randomised phase 2 study
Mark Brantly1, James Stocks2, Jorge Lascano1, Tammy Flagg1, Ann M. Jeffers2, Shuzi Z. Owens2, Torry A. Tucker2, Megan Devine2 Noga Alagem3, Naveh Tov3
1 Division of Pulmonary, Critical Care & Sleep Medicine, Department of Medicine in the College of Medicine, University of Florida, Gainesville, FL, USA
2 University of Texas Health Science Center at Tyler Center for Clinical Research, Tyler, TX, USA
3 Kamada Ltd, Rehovot, Israel.
Corresponding author: Prof. Mark Brantly, MD, Division of Pulmonary, Critical Care & Sleep Medicine, Department of Medicine in the College of Medicine, University of Florida, Gainesville, FL, USA
Tel: 352-273-8737; Email: mark.brantly@medicine.ufl.edu
Summary: A randomized phase II clinical study showed that inhaled alpha-1- antitrypsin (AAT) 80 mg/day and 160 mg/day restored lower respiratory tract protease-anti-protease homeostasis and reduced inflammatory markers in AAT- deficient individuals.
Author contributions:
Mark Brantly and Naveh Tov designed the study. Mark Brantly, Jorge Lascano, James Stocks and Meghan Devine performed the BAL procedures and sample processing. Ann Jeffers, Shuzi Owens and Tory Tucker performed sample processing,
Tammy Flagg performed laboratory analysis, Mark Brantly, Naveh Tov and Noga Alagem performed data analysis and wrote the manuscript.
Conflict of interest: Naveh Tov and Noga Alagem are Kamada employees. All other authors have no conflict of interest to declare.
Support Statement: The clinical study was supported by Kamada Ltd. Kamada personnel were involved in study design and data analysis.
Abstract
Background: Alpha-1-Antitrypsin (AAT) deficient individuals have a greater risk for developing chronic obstructive pulmonary disease than individuals with normal AAT levels.
Methods: A double-blind randomised parallel group, placebo-controlled trial to examine the safety and tolerability of “Kamada-AAT for Inhalation” (inhaled AAT) in subjects with AAT deficiency, and to explore its effect on AAT and biomarkers in the lung epithelial lining fluid (ELF). Thirty-six patients with severe AAT deficiency were randomized 2:1 to receive 80 mg or 160 mg inhaled AAT or placebo once daily for 12 weeks. The primary outcomes were AAT and anti-neutrophil elastase capacity (ANEC) in bronchoalveolar lavage (BAL) and plasma after treatment. Secondary outcomes included safety, levels of normal M-type AAT in the plasma, and concentrations of AAT, neutrophil elastase (NE), AAT-NE complexes, and neutrophil count in the ELF.
Results: Twelve weeks of active treatment significantly increased AAT, ANEC and AAT-NE complexes in the ELF. Mean antigenic AAT levels in the ELF were restored to 5.2±2.3 μM in the 80 mg arm and to 17.7 ± 2 μM in the 160 mg arm. Both doses significantly restored AAT anti-protease activity within the lung and reduced NE levels. M-specific AAT levels in plasma increased in a dose dependent manner. A clinically meaningful reduction in ELF neutrophil % was observed in the 80 mg arm. AAT for inhalation was well tolerated.
Conclusions: Inhaled AAT restores protease anti-protease homeostasis and may represent a safe and effective therapy
Funding: Kamada Ltd.
Clinical trial registration: NCT02001688
Introduction
Alpha-1 antitrypsin (AAT) inhibits neutrophil elastase (NE) activity in the lung, protecting it from proteolytic damage. In the absence of AAT, interstitial elastin is gradually degraded by NE, resulting in chronic lung disease [1].
Normal AAT levels in serum and lung epithelial lining fluid (ELF) are 20-53 µM [2] and 2-5 μM [3], respectively. The therapeutic target of AAT replacement (augmentation) therapy in individuals with AAT deficiency (AATD) is to maintain serum AAT levels above 11 μM, which is equivalent to 1.7 μM in the ELF [4-6].
This 11 μM threshold was established empirically, as validated biomarkers and dose- response data are lacking [7, 8]. Augmentation therapy was shown in an RCT to slow the rate of lung density decline [9] and by extrapolation, increase survival [10]. The survival benefit was demonstrated in one of two registry studies [11, 12]. A higher target at the physiological range may enhance treatment efficacy as measured using CT lung density [13] and inflammatory markers in bronchoalveolar lavage (BAL) fluid [14].
Augmentation treatment is currently given intravenously, and only 5-10% of serum AAT partitions to the lung. Dose-efficient approaches to augment AAT and maintain it within physiological levels at the lung are needed. Aerosolised AAT inhalation provides a more convenient administration route over intravenous AAT augmentation with potentially higher pulmonary AAT concentrations, especially in small airways and lower systemic exposure [15, 16].
Aerosolized AAT distributes to the peripheral lung [17]. Inhalation of 100 mg AAT twice daily (BID) for 7 days increased trough AAT levels in the ELF from 0.28 μM to
5.86 μM [18]. Inhalation of a single 200-mg dose of recombinant AAT increased AAT levels in the ELF 40-fold to 9 µM at 4 hours, and 5-fold at 24 hours [19].
In a phase 2/3 study that evaluated the safety and efficacy of inhaled AAT 160 mg/day in AATD patients with frequent exacerbations, inhaled AAT did not decrease the time to moderate or severe exacerbations, but a delay in the decline of forced expiratory volume in 1 second (FEV1) in the treatment arm was suggested [20].
We conducted a post-hoc analysis of a phase 2 study that examined the safety and tolerability of 80 mg/day and 160 mg/day AAT for Inhalation in subjects with AATD and explored its effect on AAT and biomarker levels in the ELF and plasma.
Methods
Patients and study design
The double-blind, randomised, parallel group, placebo-controlled trial (NCT02001688) was conducted at two sites in the USA between August 2014 and May 2016.
After approval by the sites’ institutional review boards, informed consent was obtained from men and women aged 18-65 with AATD (defined as serum AAT levels
≤11 µM with AATD genotype ZZ or Z null) and post-bronchodilator FEV1 ≥50% predicted [21]. Subjects were excluded if they had immunoglobulin A (IgA) deficiency, a history of life-threatening allergy or anaphylactic reactions, a respiratory exacerbation within the 6 weeks prior to study baseline or had actively smoked during the previous 12 months. Subjects treated with intravenous AAT at enrolment had to agree to forego this therapy for at least 8 weeks prior to initial dosing and for the entire study duration.
Eligible patients were randomised 2:1 separately at each site to receive Kamada-AAT for Inhalation (Kamada Ltd., Rehovot, Israel) or placebo. A randomization list was generated for each site separately. Packaging of Active drug and placebo were identical to maintain blinding.
Treatment
Kamada-AAT for Inhalation or placebo were administered with an eFlow® nebulizer (PARI Pharma GmbH, Munich, Germany). Subjects at site 1 received a single daily inhalation (4 ml solution) of AAT 80 mg or placebo. At site 2, subjects received AAT 80 mg BID (i.e., a total of 160 mg/day) or placebo. After a 12-week double-blind treatment period, all subjects regardless of initial treatment allocation were invited to participate in an open-label extension (OLE) where they received AAT 80 mg BID for 12 additional weeks.
Outcomes
The primary study endpoints were the levels of antigenic and functional AAT in ELF of subjects treated with AAT versus placebo.
The secondary endpoints were safety and tolerability of Kamada-AAT for Inhalation (determined by adverse event rate), plasma M-type AAT (PiM) levels, AAT concentrations, NE, AAT-NE complexes, and neutrophil count in the ELF. Lung function was evaluated by FEV1.
Exploratory endpoints were anti-drug antibodies (ADA), and ELF inflammatory marker levels (interleukin [IL]-6, IL-8, macrophages cell counts, eosinophils, lymphocytes, and neutrophils).
Procedures
BAL was performed at baseline (three days before treatment initiation) and 12±4 (site 2) or 24±4 (site 1) hours after the last dose of the double-blind period. The timing relative to the last dose was set to capture trough drug levels. 3-4 lobes were sampled per patient. 100 mL saline was instilled into each sampled lobe. If BAL retrieval was less than 20%, the sample wasdeemed low quality and excluded from analysis. The final analyte concentration, corrected per urea concentration in the sample and plasma, was determined separately for each sampled lobe, and the results from all sampled lobes were averaged per subject. BAL was not performed at the end of the OLE. See the supplementary material for analyte assay methods.
Statistical analysis
The study hypothesis postulated that subjects treated with inhaled AAT would have higher levels of AAT and anti-NE capacity (ANEC) in the ELF versus placebo, but the study was not powered for any formal hypothesis testing.
All subjects who received at least one dose of inhaled AAT or placebo were included in the analysis. Changes from baseline to week 12 in antigenic and functional AAT levels in ELF were analysed using the non-parametric Wilcoxon signed-rank test (for within group changes) and the non-parametric Wilcoxon rank-sum test and Kruskal- Wallis test (for changes between arms). Dose dependence was evaluated using Mann- Whitney-Wilcoxon rank-sum test (2 groups: 80 mg/day and 160 mg/day).
A one-sided test was used for antigenic and functional AAT levels to maintain an overall 2.5% level of significance. Otherwise, all statistical tests were two-sided with 5% significance levels.
Results
Thirty-six subjects were randomised to receive AAT or placebo (Figure 1). After completing the 12-week double-blind period, 26 subjects (10 and 8 subjects in the AAT 80 mg and 160 mg arms, respectively and 8 subjects in the placebo arm) enrolled in the OLE, and received AAT 160 mg/day for 12 weeks. Baseline demographic characteristics and FEV1 predicted values were comparable among the treatment arms (Table 1).
AAT levels in ELF
After 12 weeks of treatment, mean antigenic AAT and ANEC levels measured in the ELF of subjects in the 80 mg and 160 mg arms statistically significantly increased from baseline, whereas no statistically significant changes were observed in the pooled placebo arm (Table 2 and Figure 2 A,B). After 12 weeks of treatment, ANEC levels were linear with antigenic AAT levels in both treatment arms (Figure 2 C,D).
Antigenic AAT levels in the ELF were dose dependent with an estimated difference of 7838.4 nM (95% CI, 1531.1-14997.2 nM, p=0.019) between the medians of the 80 mg and the 160 mg arms. The median ANEC levels were numerically higher in the 160 mg arm compared to the 80 mg arm, but the difference was not statistically significant; estimated difference 1408.0 nM (95% CI, (−)982.9-6127.1 nM; p=0.249). The study met its primary endpoint of significant change from baseline in antigenic and functional AAT in the pooled treatment arms vs. the pooled placebo arms (p<0.0001 for both).
AAT Activity in the ELF
Only 4/18 patients in site 1 and 3/18 in site 2 had detectable AAT-NE complexes in ELF at baseline (mean AAT-NE 7.5±2.7 nM). After 12 weeks of treatment, AAT-NE complexes were detected in 12/12 patients in the 80 mg arm, (mean 37.6 nM±27.9 nM),
10/11 patients in the 160 mg arm (154.9±237.2 nM) and in 1/11 patients in the placebo arm (6.9 nM). At week 12, AAT-NE levels were statistically significantly higher in the 160 mg arm vs the 80 mg arm (p=0.028).
Neutrophil inflammatory markers in the ELF
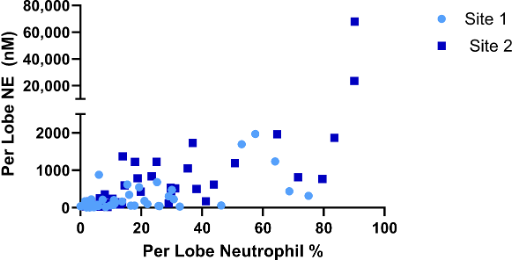
Baseline mean NE levels and neutrophil counts varied highly among subjects (Figure 3), and even among lobes in the same subject. To control for variability, inflammatory markers were analysed only in lobes that were sampled at baseline and resampled at 12 weeks (Supplementary Table 1). At baseline, mean neutrophil differentials ranged from 0 to 90%. At site 1, baseline neutrophil % ranged from 0 to 75% and at site 2, from 1.8 to 90.2 %. Mean NE levels increased steadily with neutrophil percentage up to 80%.
Two outlier lobes, which were sampled from one patient in the placebo arm at site 2, had a neutrophil percentage above 80% and NE levels higher by an order of magnitude than all other measurement (Supplementary Figure 1). These lobes were excluded from the NE analysis. The patient’s medical history indicated bronchiectasis, and it was assumed that the high observed inflammatory load could be attributed to a bronchiectasis flare-up. A third lobe from that patient with differential neutrophil counts < 80% was not excluded from analysis.
Mean NE levels statistically significantly decreased from baseline to week 12 in both treatment arms. A non-statistically significant decrease from baseline in mean NE levels was observed in the placebo arm (Figure 3).
Mean neutrophil percentages were generally higher than normal at baseline (Table 3). Baseline mean neutrophil percentages were higher in site 2 vs site 1, and were highest among the placebo group in site 2. After 12 weeks of treatment with inhaled AAT, mean neutrophil percentage decreased significantly and macrophage percents increased
significantly in the 80 mg/day arm, but not in the 160 mg/day arm nor in the placebo arm. Mean eosinophils and lymphocyte percentages were generally similar among all arms and did not change appreciably between baseline and week 12.
M-Type AAT in plasma
After 12 weeks of treatment median PiM statistically significantly increased from baseline by 119 nM (95% CI, 60-162; p<0.0002) and 101 nM (95% CI, 35-370;
p<0.0018), in the 80 mg and 160 mg arms, respectively. Median PiM levels were similar in both treatment arms, with a higher range in the 160 mg arm (Figure 4A). PiM levels were dose dependent and showed linearity with ELF AAT levels up to ~20 µM. At supraphysiological levels (>20 µM), plasma PiM levels were lower than expected. After 12 weeks of treatment with AAT 160 mg/day in the OLE, subjects who received placebo for the study’s first 12 weeks, had mean PiM levels of 135 nM (range 89-452 nM).
Pulmonary function
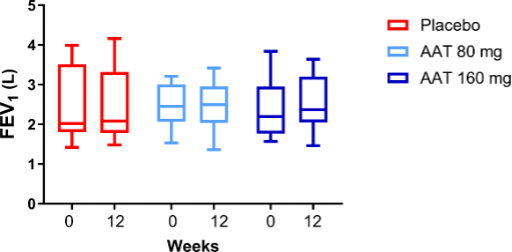
Mean FEV1 predicted remained within ±5% of pre-dose levels at all time points in all treatment arms (Supplementary Figure 3)
Safety and tolerability
During the double-blind period, 83.3% (10/12), 66.7% (8/12) and 58.3% (7/12) of subjects in the 80 mg, 160 mg, and pooled placebo arm had at least one TEAE. The proportion of subjects who experienced at least one treatment-related TEAE was similar across treatment groups (41.7% [5/12], 33.3% [4/12] and 41.7% [5/12] in the 80 mg, 160 mg, and placebo arms, respectively). One subject in the 160 mg AAT group had serious pneumonia, which was considered unrelated to the study treatment but led to discontinuation of the study drug. Cough was the most commonly reported drug-related
TEAE in all three treatment groups (33.3% [4/12], 8.3% [1/12] and 25.0% [3/12] of subjects in the 80 mg, 160 mg, and placebo arms, respectively). Fifteen subjects (57.7%) had TEAEs during the OLE (none were treatment-related).
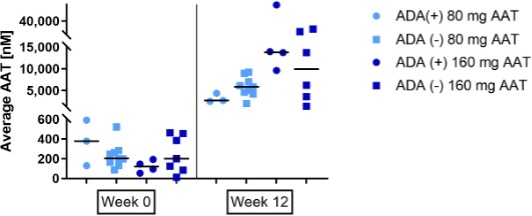
Twelve of 32 subjects (38%) exposed to AAT either during the double-blind or the OLE periods developed ADAs. All binding antibody titres were equal or lower than 1:16 except for one subject, with a titre of 1:32. All patients with ADAs had neutralizing ADAs. No impact on safety was observed (including no classical anaphylactic or hypersensitivity reactions), nor any impact on AAT concentrations in the ELF or plasma (Supplementary Figure 2).
Follow-up plasma samples were collected from 25 of the 32 exposed patients, at up to 48 months after the last dose. 11 of the 12 seropositive subjects provided follow-up samples, and 10/11 (91%) no longer had detectable ADAs. The patient who was still seropositive at follow-up had an ADA titre of 4 versus 8 during the study with no neutralizing antibodies. Since the end of the study, this patient reported receiving intravenous AAT and had no outstanding health issues or hospitalizations. All patients who were ADA-negative during the study remained negative at follow-up.
Discussion
This study demonstrated that 12 weeks of treatment with AAT for inhalation 80 mg/day or 160 mg/day significantly increased functional AAT levels as evidenced by the increase in ANEC and AAT-NE complexes in the ELF. Both doses restored AAT anti-protease activity within the lung, with a concomitant decrease in ELF neutrophilic burden, evidenced by a significant decrease in NE. These results suggest that aerosol AAT replacement is highly effective in relieving the elastolytic burden in the lower airways of AATD patients.
The baseline values of antigenic AAT measured in in the ELF (~200 nM) illustrate the severe AAT deficiency of study participants. Baseline ANEC levels were higher than antigenic AAT levels, reflecting the presence of additional serine protease inhibitors in the lower airways [22, 23]. Inhalation of AAT 80 mg/day restored mean antigenic AAT trough levels in the ELF to the upper range of normal physiological levels, while inhalation of AAT 160 mg/day restored them to well above the normal physiological range in most patients. These values are higher than those reported for intravenous AAT augmentation [7, 24]. Notably, AAT levels were restored in all sampled lobes.
Inflammatory markers were highly variable among patients, but also within different lobes in the same patient. This observation underlies substantial heterogeneity in pulmonary inflammation among different areas in the lung. The original analysis plan for the study dictated that acceptable results (i.e., BAL return >20%) from all sampled lobes will be averaged per subject. This plan contributed to variability of intra-subject inflammatory marker levels in cases where lobes were not revisited or results were unacceptable at random. Analysing per-lobe data provided a plausible solution and was useful for describing the anti-neutrophilic effects of AAT for inhalation, especially in the 160 mg arm site where the inflammatory load was highly variabile . The results of this study highlight the importance of revisiting pulmonary lobes in longitudinal BAL studies. We further suggest that chest imaging should be considered before BAL procedures to locate highly emphysematous or bronchiectatic regions to better consider their contribution to the inflammatory load and to drug effects.
Although NE decreased significantly in both treatment arms, neutrophil percent decreased only in the 80 mg arm. Interestingly, baseline disease was more advanced at site 2, as evidenced by FEV1%, NE and neutrophil%. Further studies are required
to determine whether AAT replacement by inhalation is most beneficial if applied very early in the disease course. Since mild pulmonary inflammation is already present in AATD before lung function starts to deteriorate [25], early intervention may modify the disease and its outcomes.
PiM levels in subjects treated with inhaled AAT 80 mg or 160 mg, but not placebo, increased significantly from baseline, indicating diffusion of inhaled AAT across the pulmonary interstitium into the blood. PiM concentrations (100-300 nM), however, were below the targeted plasma level for intravenous augmentation (11 μM).
Endogenous or augmented ELF AAT levels are typically 10% of those in serum [5], however 10% partition is not expected for delivery by inhalation since inhaled AAT is diluted into the plasma. Considering adult plasma volume to be 3.5 L, PiM concentrations of ~150 nM (0.00815 mg/mL) are equivalent to ~28.5 mg, or 35% of the daily dose. Deposition of AAT for inhalation in the lower airways of non-AATD COPD patients was found to be ~40% of the daily dose in an early phase study (unpublished). Therefore, PiM levels after 12 weeks of active treatment are roughly equivalent to the deposited daily dose, suggesting that AAT for inhalation traverses the pulmonary interstitium. However, Inhaled AAT was not designed to treat rare systemic manifestations of AATD, such as panniculitis; based on this study’s results, IV treatment is best suited in these cases.
Inhaled AAT showed a good safety profile. FEV1 was stable throughout the study, suggesting that inhaled AAT does not compromise pulmonary function.
The lung is a major hub of new antigen exposure, and this is probably that reason that 12 patients (29%) who were treated with AAT for inhalation developed ADAs. The antibody response demonstrated a generally low titre (mostly 1:16 and below). No
hypersensitivity or anaphylactoid reactions were observed and no adverse events were associated with an antibody-positive status. Importantly, plasma and ELF AAT levels were unaffected by antibody presence. Follow-up of the ADA-positive subjects showed that antibodies were no longer detectable in 91% of them 48 weeks after the last inhaled dose. The one subject who still had detectable antibodies had a low titre with no detectable neutralizing antibodies.
The study limitations include its small sample size per treatment group and relatively short treatment period. Site variations in BAL technique were not optimally controlled. The time between the last dose of inhaled AAT and BAL was different between dose arms. This is due to the study being designed to capture the steady-state trough AAT concentration. This timing difference precludes conclusively assigning the observed differences between treatment arms to dose alone. However, the linear relationship between AAT and ANEC in the ELF, and between PiM and ELF AAT reassures the overall validity of the results.
This phase II study is the largest to date where BAL was used to evaluate ELF content in AATD. It is also the first to show that AAT inhalation is a safe and efficient modality for delivering AAT to the airways. AAT at a dose of 80 mg/day was found to be adequate to restore lung anti-proteolytic activity with higher dose efficiency compared to the approved intravenous AAT dose of 60 mg/kg/week. A phase 3 study (NCT04204252) investigating the safety and efficacy of AAT for inhalation 80 mg/day is ongoing.
Table 1. Demographic and baseline characteristics of the intent to treat population
| Parameter | 80 mg/day N=12 | 160 mg/day N=12 | Placebo N=12 | P value |
| Age, years, mean (SD) | 55.5 (8.7) | 55.3 (10.1) | 56 (4.8) | 0.706a |
| Gender Female/Male, n | 9/3 | 7/5 | 8/4 | 1.0000b |
| Body mass index, kg/m2, mean (SD) | 30.0 (4.3) | 28 (8.3) | 26 (4.8) | 0.1180a |
| Lung function | ||||
| FEV1, L, mean (SD) | 2.5 (0.5) | 2.4 (0.7) | 2.5 (0.9) | 0.8760a |
| FEV1, % of predicted, mean (SD) | 81.7 (17.9) | 73.2 (20.1) | 78.1 (20.0) | 0.9232a |
| Plasma measurements | ||||
| AAT, mg/dL, mean (SEM) | 4.5 (0.3) | 4.8 (0.3) | 4.7 (0.3) | 0.9954c |
| M-Type AAT, nM, mean (SD) | 26 (21) | 38 (45) | 23 (21) | 0.2629c |
AAT, alpha-1 antitrypsin; FEV1, forced expiratory volume in one second; FVC, forced vital capacity; SD, standard deviation; SEM, standard error of the mean. The following tests compared pooled treatment vs. Placebo. A- Unpaired t test, b- Fisher’s exact test c- Mann Whitney (exact) test
Table 2. Change in AAT levels in ELF and plasma at baseline and 12 weeks
| 80 mg AAT/day | 160 mg AAT/day | Pooled placebo | ||||||||
| Week | 0 | 12 | P value | 0 | 12 | P value | 0 | 12 | P value | |
| ELF | N | 12 | 12 | 12 | 10 | 12 | 10 | |||
| Antigenic AAT, µM, mean (SD) | 0.27 (0.16) | 5.23 (2.35) | 0.0005 | 0.20 (0.15) | 17.76 (20.02) | 0.002 | 0.20 (0.11) | 0.23 (0.16) | 1.0 | |
| ANEC, µM, Mean (SD) | 1.43 (1.26) | 4.01 (1.51) | 0.0005 | 1.23 (1.56) | 6.65 (6.85) | 0.0039 | 0.99 (0.72) | 1.08 (0.75) | 1.0 | |
| Plasma | N | 11 | 12 | 12 | 11 | 12 | 11 | |||
| M-Type AAT, nM, Mean (SD) | 26 (21) | 137 (60) | 0.0010 | 38 (45) | 202 (143) | 0.0020 | 23 (21) | 15 (8) | 0.2783 | |
AAT, alpha-1 antitrypsin; ANEC, anti-NE capacity; ELF, epithelial lining fluid; IQR, interquartile range; SD, standard deviation; SEM, standard error of the mean. ELF Raw data was measured in nM, results are given in micromolars, rounded to two digits after the decimal. PiM results were rounded to the digit. P value was calculated using Wilcoxon matched-pairs signed rank test.
Table 3. Inflammatory cell counts in ELF at week 0 (baseline) and after 12 weeks of treatment with AAT (80 or 160 mg/day) or placebo.
| 80 mg AAT/day N=11 L=27 | Placebo Site 1 N=5 L=12 | 160 mg AAT/day N=10 L=17 | Placebo Site 2 N=5 L=9 | ||||||||||
| Week | 0 | 12 | P | 0 | 12 | P | 0 | 12 | P | 0 | 12 | P | |
| Neutrophils % | Media n (CI) | 8.3 (4.3,22) | 5.5 (2,13) | 0.005 3 | 11 (1.5,25) | 5 (1.9,53) | 0.965 8 | 13.3 (6.9,17.4) | 10.6 (3.7,27.6) | >0.999 9 | 71.6 (6.2,90.1) | 15.5 (12.9,73) | 0.203 1 |
| Mean (SD) | 17 (19) | 10 (13) | 17 (19) | 20 (28) | 16 (13) | 17 (19) | 51 (38) | 36 (30) | |||||
| Lymphocytes % | Media n (CI) | 5.8 (4.3,9.4) | 5.3 (2.8,8) | 0.786 4 | 1 (0.2,6.6) | 2.9 (1,24) | 0.077 1 | 7 (2.5,9) | 4.3 (1.6,7.3) | 0.5554 | 2.6 (1.3,5.7) | 3.5 (1.2,6.2) | 0.593 8 |
| Mean (SD) | 7.3 (7) | 6.3 (5.2) | 5.7 (9.5) | 11 (15) | 6.5 (4) | 5.9 (5.2) | 3.1 (2.4) | 4.4 (2.9) | |||||
| Macrophages % | Media n | 82 (72,87) | 87 (76,92) | 0.012 1 | 81 (55,98) | 77 (43,91) | 0.124 | 80 (73,84) | 79 (70,88) | 0.9632 | 27 (7,90) | 76 (26,81) | 0.203 1 |
| (CI) | |||||||||||||
| Mean (SD) | 75 (21) | 83 (13) | 77 (21) | 68 (25) | 76 (11) | 76 (16) | 46 (37) | 59 (29) | |||||
| Eosinophils % | Media n (CI) | 0 (0,0) | 0 (0,0.4) | 0.492 9 | 0 (0,1.1) | 0.4 (0,0.8) | 0.863 3 | 0.3 (0,1.1) | 0 (0,1) | 0.5527 | 0.6 (0,1.3) | 0.5 (0,1.2) | 0.414 1 |
| Mean (SD) | 0.2 (0.5) | 0.4 (0.5) | 0.6 (1.2) | 0.5 (0.6) | 0.7 (0.8) | 0.5 (0.7) | 0.7 (0.5) | 0.5 (0.6) |
Data was analyzed per-lobe, using lobes that were sampled both at baseline and at week 12 with bronchoalveolar lavage return over 20%. N, number of patients, L number of lobes that contributed to analysis. P, the P- value of Wilcoxon matched pairs sign-ranked test for the change from baseline to week 12. SD, standard deviation, CI, 95% confidence interval of median
References
- Kalfopoulos M, Wetmore K, ElMallah MK. Pathophysiology of Alpha-1 Antitrypsin Lung Disease. Methods Mol Biol 2017: 1639: 9-19.
- Brantly ML, Wittes JT, Vogelmeier CF, Hubbard RC, Fells GA, Crystal RG. Use of a highly purified alpha 1-antitrypsin standard to establish ranges for the common normal and deficient alpha 1-antitrypsin phenotypes. Chest 1991: 100(3): 703-708.
- Hubbard RC, Crystal RG. Strategies for aerosol therapy ofα 1-antitrypsin deficiency by the aerosol route. Lung 1990: 168(1): 565-578.
- Sandhaus RA, Stocks J, Rouhani FN, Brantly M, Strauss P. Biochemical efficacy and safety of a new, ready-to-use, liquid alpha-1-proteinase inhibitor, GLASSIA (alpha1-proteinase inhibitor (human), intravenous). COPD 2014: 11(1): 17-25.
- Wewers MD, Casolaro MA, Crystal RG. Comparison of alpha-1-antitrypsin levels and antineutrophil elastase capacity of blood and lung in a patient with the alpha-1-antitrypsin phenotype null-null before and during alpha-1-antitrypsin augmentation therapy. Am Rev Respir Dis 1987: 135(3): 539-543.
- Wewers MD, Casolaro MA, Sellers SE, Swayze SC, McPhaul KM, Wittes JT, Crystal RG. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. The New England journal of medicine 1987: 316(17): 1055-1062.
- Franciosi AN, Fraughen D, Carroll TP, McElvaney NG. Alpha-1 antitrypsin deficiency: clarifying the role of the putative protective threshold. Eur Respir J 2021.
- Lopez-Campos JL, Carrasco Hernandez L, Caballero Eraso C. Implications of a Change of Paradigm in Alpha1 Antitrypsin Deficiency Augmentation Therapy: From Biochemical to Clinical Efficacy. J Clin Med 2020: 9(8).
- Chapman KR, Burdon JG, Piitulainen E, Sandhaus RA, Seersholm N, Stocks JM, Stoel BC, Huang L, Yao Z, Edelman JM, McElvaney NG, Group RTS. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet (London, England) 2015: 386(9991): 360-368.
- McElvaney NG, Burdon J, Holmes M, Glanville A, Wark PAB, Thompson PJ, Hernandez P, Chlumsky J, Teschler H, Ficker JH, Seersholm N, Altraja A, Mäkitaro R, Chorostowska-Wynimko J, Sanak M, Stoicescu PI, Piitulainen E, Vit O, Wencker M, Tortorici MA, Fries M, Edelman JM, Chapman KR. Long-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). The Lancet Respiratory Medicine 2017: 5(1): 51-60.
- Fraughen DD, Ghosh AJ, Hobbs BD, Funk GC, Meischl T, Clarenbach CF, Sievi NA, Schmid- Scherzer K, McElvaney OJ, Murphy MP, Roche AD, Clarke L, Strand M, Vafai-Tabrizi F, Kelly G, Gunaratnam C, Carroll TP, McElvaney NG. Augmentation Therapy for Severe Alpha-1 Antitrypsin Deficiency Improves Survival and Is Decoupled from Spirometric Decline-A Multinational Registry Analysis. Am J Respir Crit Care Med 2023: 208(9): 964-974.
- Ellis PR, Holm KE, Choate R, Mannino DM, Stockley RA, Sandhaus RA, Turner AM. Quality of Life and Mortality Outcomes for Augmentation Naive and Augmented Patients with Severe Alpha-1 Antitrypsin Deficiency. Chronic Obstr Pulm Dis 2023: 10(2): 139-147.
- Tortorici MA, Rogers JA, Vit O, Bexon M, Sandhaus RA, Burdon J, Chorostowska- Wynimko J, Thompson P, Stocks J, McElvaney NG, Chapman KR, Edelman JM. Quantitative disease progression model of alpha-1 proteinase inhibitor therapy on computed tomography lung density in patients with alpha-1 antitrypsin deficiency. Br J Clin Pharmacol 2017: 83(11): 2386-2397.
- Campos MA, Geraghty P, Holt G, Mendes E, Newby PR, Ma S, Luna-Diaz LV, Turino GM, Stockley RA. The Biological Effects of Double-Dose Alpha-1 Antitrypsin Augmentation Therapy. A Pilot Clinical Trial. Am J Respir Crit Care Med 2019: 200(3): 318-326.
- Hubbard RC, Crystal RG. Strategies for aerosol therapy of alpha 1-antitrypsin deficiency by the aerosol route. Lung 1990: 168 Suppl: 565-578.
- Kerem E, Bauer S, Strauss P, Jaffe N, Armoni S, Pugatsch T, Shoseyov D, Tov N. Safety and Efficacy of Inhaled Human Alpha-1 Antitrypsin (AAT) in Cystic Fibrosis (CF): A Report of a Phase II Clinical Study. A26 NEW DATA REGARDING CYSTIC FIBROSIS. Am Thoracic Soc, 2009; p. A1185.
- Kropp J, Wencker M, Hotze A, Banik N, Hubner GE, Wunderlich G, Ulbrich E, Konietzko N, Biersack HJ. Inhalation of [123I]alpha1-protease inhibitor: toward a new therapeutic concept of alpha1-protease inhibitor deficiency? J Nucl Med 2001: 42(5): 744-751.
- Hubbard RC, Brantly ML, Sellers SE, Mitchell ME, Crystal RG. Anti-neutrophil-elastase defenses of the lower respiratory tract in α1-antitrypsin deficiency directly augmented with an aerosol of α1-antitrypsin. Annals of internal medicine 1989: 111(3): 206-212.
- Hubbard RC, McElvaney NG, Sellers SE, Healy JT, Czerski D, Crystal R. Recombinant DNA- produced alpha 1-antitrypsin administered by aerosol augments lower respiratory tract antineutrophil elastase defenses in individuals with alpha 1-antitrypsin deficiency. Journal of Clinical Investigation 1989: 84(4): 1349.
- Stolk J, Tov N, Chapman KR, Fernandez P, MacNee W, Hopkinson NS, Piitulainen E, Seersholm N, Vogelmeier CF, Bals R, McElvaney G, Stockley RA. Efficacy and safety of inhaled alpha-1-antitrypsin in patients with severe alpha-1-antitrypsin deficiency and frequent exacerbations of Chronic Obstructive Pulmonary Disease. Eur Respir J 2019.
- Quanjer PH, Cooper B, Ruppel GL, Swanney MP, Stocks J, Culver BH, Thompson BR. Defining airflow obstruction. Eur Respir J 2015: 45(2): 561-562.
- Vogelmeier C, Hubbard RC, Fells GA, Schnebli HP, Thompson RC, Fritz H, Crystal RG. Anti- neutrophil elastase defense of the normal human respiratory epithelial surface provided by the secretory leukoprotease inhibitor. J Clin Invest 1991: 87(2): 482-488.
- Greene CM, McElvaney NG. Proteases and antiproteases in chronic neutrophilic lung disease – relevance to drug discovery. Br J Pharmacol 2009: 158(4): 1048-1058.
- Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest 1981: 68(5): 1158-1165.
- Kokturk N, Khodayari N, Lascano J, Riley EL, Brantly ML. Lung Inflammation in alpha-1- antitrypsin deficient individuals with normal lung function. Respir Res 2023: 24(1): 40.
Figure legends
Figure 1. CONSORT diagram of the study’s intent to treat population
Figure 2: Levels of antigenic AAT (A) and anti-neutrophil elastase capacity (B) in the ELF at baseline and after 12 weeks of treatment with AAT for Inhalation or placebo. Data points are the average value obtained from separate lobes. The dotted lines show the normal range. P values are indicated where differences were statistically significant. a P value for the change from baseline to 12 weeks was calculated using the (Exact) Wilcoxon signed-rank test; b P value for the difference between the 80 mg/day and the 160 mg/day arms was calculated using the Mann-Whitney-Wilcoxon rank-sum test (2 groups). C and D. show Per-lobe ANEC and AAT at week 12 in the 80 mg arm and placebo (C) and in the 160 mg arm (D). The straight line is a linear regression through the treatment arm values.
Figure 3. Neutrophil Elastase in the ELF at baseline and after 12 weeks of treatment with AAT for Inhalation or placebo. Lines depict the median. P values are indicated where differences were statistically significant. The p value for the change in ELF NE from baseline to 12 weeks in each treatment arm (bottom) was calculated using the (Exact) Wilcoxon signed-rank test.
Figure 4. M-specific AAT (PiM) measured in plasma. (A) PiM levels at baseline and after 12 weeks treatment with AAT for Inhalation or placebo. Horizontal lines depict the median. P values are indicated where differences from baseline within treatment groups were statistically significant. P values were calculated using the (Exact) Wilcoxon
signed-rank test. The treatment effect was also statistically significant vs. placebo for both arms. B Average ELF antigenic AAT vs. plasma PiM at week 12 in patient samples. The solid line is a linear regression using pooled data from 80 mg and 160 mg treatment arms. Two outliers marked with an asterisk were excluded from the regression analysis.
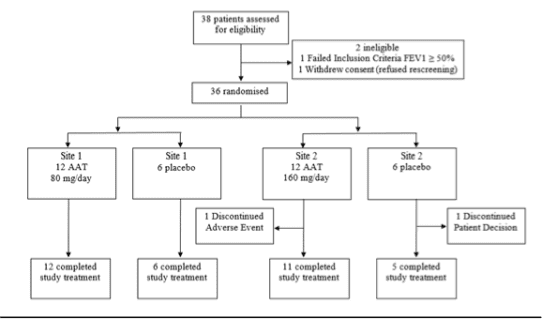
Figure 1
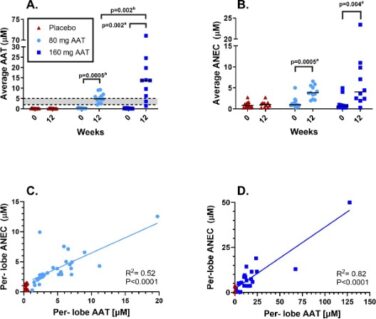
Figure 2
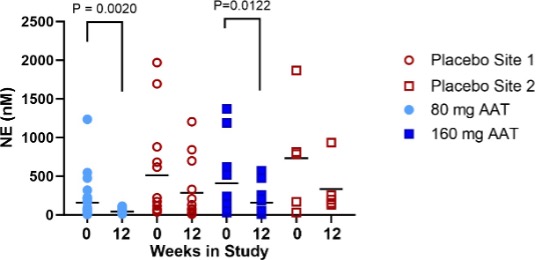
Figure 3
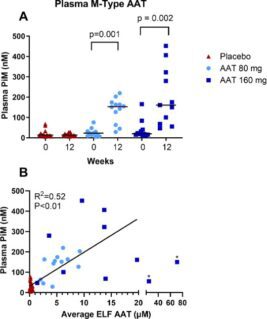
Figure 4
Supplementary materials
M-AAT-specific ELISA
M-type alpha-1-antitrypsin (AAT) was measured by enzyme-linked immunosorbent assay (ELISA) as previously described [1] using a monoclonal antibody (mAb) developed in a standard fashion by injecting mice with highly purified PI M1M1 AAT. Clones that made specific antibodies to M- but not to Z-AAT were identified in a dot blot assay. Dilutions of serum were added to wells coated with this mAb at 167 ng/mL, and antigen–antibody complexes were detected using a polyclonal rabbit anti- human AAT at 20 ng/mL followed by a peroxidase-conjugated goat anti-rabbit Ig, with o-phenylenediamine added for colorimetric analysis. The lower limit of quantification for this assay is 8.8 pM, with selectivity and reproducibility at the lower limit of detection >90% [1].
Anti-drug antibodies
Plasma samples were analysed using a validated semi-quantitative electrochemiluminescence bridging assay for the detection of AAT-reactive immunoglobulin G (IgG) antibodies developed according to guidelines by Charles River Laboratories Preclinical Services (Montreal, Canada). Briefly, serum IgG was purified on a Protein G spin column and then incubated with AAT conjugated to ruthenium to allow formation of ruthenium-labelled AAT-anti-drug antibody (ADA) complexes. The mixture was then added to electrochemiluminescence plates (Meso Scale Diagnostics, Rockville, MD, USA) preincubated with AAT conjugated to biotin. ADAs crosslinked the ruthenium and biotin and was detected by electrochemiluminescence [2].
Determination of antigenic AAT levels in epithelial lining fluid (ELF)
AAT concentration in bronchoalveolar lavage (BAL) fluid was determined by indirect sandwich ELISA. Briefly, Immulon-2 plates (Dynatech, Chantilly, VA, USA) were coated overnight with goat anti-human AAT antibody (Capple, Organon Teknika Corp., Durham, NC, USA). Plates were washed, and samples and standards were added. Purified AAT from normal plasma was used to generate a standard curve [3]. The plates were washed and incubated at Temp °C with a rabbit anti-human AAT antibody (Boehringer Mannheim Biochemicals, Indianapolis, IN, USA) followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG (Boheringer Mannheim Biochemicals, Indianapolis, IN, USA). Finally, o-phenylendiamine-dihydrochoride substrate (Fluka Biochemika, Milwaukee, WI, USA) was added to develop the ELISA. To standardize ELF measurements taken from each subjects, an ELF dilution factor was calculated using the ratio of plasma urea concentration to bronchoalveolar lavage fluid (BALF) urea concentration. BALF measurements were multiplied were this factor.
Determination of functional AAT levels in BAL Fluid (anti-neutrophil elastase capacity [ANEC] assay)
All BALF samples were treated with 4M methylamine (Sigma, St.Louis, MO, USA) at room temperature for 1 hour. Standards and samples were added to the plates and these were incubated at 37°C for 5 minutes. Human neutrophil elastase (NE) was added and incubated for 5 minutes (Athens Research and Technology Inc., Athens, GA, USA). Methoxysuccinyl-ala-ala-val-pro-p-nitroanilide (Sigma) was added and kinetic optical density at 405-490 nm was determined.
Determination of AAT:NE complex concentrations in BALF
Plates were coated with sheep anti-human NE (ICN, Costa Mesa, CA, USA) and incubated at 4°C overnight. Plates were washed and samples and standards loaded for incubation at 37°C for 1.5 hours. Rabbit anti-AAT antibody was added and incubated at 37°C for 1.5 hours. The plates were then washed and horseradish peroxidase conjugated goat anti-rabbit IgG was added. Reactions were terminated by H2SO4, and the optical density at 490 nm was determined. Values were corrected for BALF dilution to give ELF concentrations of AAT:NE complexes.
Determination of antigenic NE concentrations
Samples and standards pre-treated with 1 mM phenylmethylsulfonyl fluoride (PMSF; (Sigma) were loaded to sheep anti-human NE-coated plates and incubated at 37°C for 1 hour. Rabbit anti-NE (Athens Research and Technology,) was added and incubated for 1 hour at 37°C. The plates were washed and horseradish peroxidase conjugated goat anti-rabbit IgG was added. Reactions were terminated by adding H2SO4, and optical density at 490 nm was determined using a SPECTRAmax (Molecular Devices, Sunnyvale, CA, USA). Values were corrected for BALF dilution to give ELF concentrations of NE.
Determination of inflammatory mediators in BALF
Cytokine levels were measured using ELISA kits (R&D Systems Minneapolis, MN, USA) according to the manufacturer’s instructions. Briefly, samples were diluted in assay buffer before being applied to the assay plates for incubation. The plates were then washed, and conjugated antibodies were applied for the second incubation. Plates were washed and the substrate was added. The optical density was measured and sample concentrations were calculated based on the standard curve.
Supplementary Table 1: Revisited lobes with BAL return >20% at each site
| Site 1 N=18 | Site 2 N=18 | |||
| Lobe | Treatment n=11 | Placebo n=5 | Treatment n=10 | Placebo n=5 |
| Lingula | 9 | 4 | 6 | 3 |
| Right Middle Lobe | 8 | 3 | 2 | 1 |
| Right Upper Lobe | 10 | 5 | 7 | 4 |
| Right Lower Lobe | 0 | 0 | 2 | 1 |
| Left Lower Lobe | 0 | 0 | 0 | 0 |
| Left Upper Lobe | 0 | 0 | 1 | 0 |
| Total | 27 | 12 | 17 | 9 |
N indicates the number of patients and n indicates the number of revisited lobes
Supplementary Table 2: Summary of Antigenic AAT by Lobe Site 1.
| 80 mg/day | Site 1 Placebo | |||||||
| AAT [nM] Median (Min, Max) | Week 0 | Week 12 | Week 0 | Week 12 | ||||
| Lobe | N | N | N | N | ||||
| LING | 11 | 232 (42, 355) | 9 | 2875 (1541, 5581) | 5 | 179 (103, 268) | 5 | 318 (183, 525) |
| RML | 10 | 199 (67, 486) | 9 | 4940 (2044, 11193) | 4 | 178 (55, 746) | 3 | 394 (307, 516) |
| RUL | 10 | 253 (116, 1097) | 10 | 6381 (1960, 19757) | 5 | 295 (79, 530) | 5 | 317 (180, 590) |
LING- Lingula, RML- Right Middle Lobe, RUL- Right Upper Lobe, LLL- Left lower lobe, LUL- Left Upper Lobe, RLL- Right Lower Lobe N- number of analyzable samples. Results were rounded to the digit.
Supplementary Table 3: Summary of Antigenic AAT by Lobe Site 2.
| 160 mg/day | Site 2 Placebo | |||||||
| AAT [nM] Median (Min, Max) | Week 0 | Week 12 | Week 0 | Week 12 | ||||
| Lobe | N | N | N | N | ||||
| LING | 8 | 210 (55, 405) | 6 | 13607 (1467, 19728) | 4 | 165 (128, 263) | 3 | 91 (12, 189) |
| RML | 7 | 126 (37, 784) | 2 | 10945 (10117, 11773) | 3 | 189 (84, 204) | 1 | 11 (11, 11) |
| RUL | 8 | 188 (52, 416) | 6 | 21182 (2810, 127531) | 5 | 166 (71, 178) | 2 | 187 (180, 195) |
| LLL | 2 | 328 (234, 423) | – | – | 1 | 132 (132, 132) | – | – |
| LUL | 2 | 337 (186, 487) | – | – | 1 | 181 (181, 181) | – | – |
| RLL | 2 | 240 (153, 328) | 2 | 9190 (1433, 16948) | 1 | 92 (92, 92) | – | – |
LING- Lingula, RML- Right Middle Lobe, RUL- Right Upper Lobe, LLL- Left lower lobe, LUL- Left Upper Lobe, RLL- Right Lower Lobe N- number of analyzable samples. Results are given in nM and were rounded to the digit.
Supplementary Figure 1: Neutrophil elastase and Neutrophil Percents at Baseline
Supplementary Figure 1: Average AAT levels in ELF of patients exposed to AAT seropositive or seronegative to anti drug antibodies, at week 0 and week 12 of the study. The horizontal bar is the median.
Supplementary Figure 3. Mean values for percent predicted FEV1 at pre-dose were comparable across all treatment arms, including placebo. Following 12 weeks of treatment, FEV1 remained within ±5% from pre-dose levels at all time points for all treatment arms
References for Supplementary Section
Brantly ML, Wittes JT, Vogelmeier CF, Hubbard RC, Fells GA, Crystal RG. Use of a highly purified alpha 1-antitrypsin standard to establish ranges for the common normal and deficient alpha 1-antitrypsin phenotypes. Chest 1991: 100(3): 703-708.
Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT, Rouhani F, Conlon TJ, Calcedo R, Betts MR, Spencer C, Byrne BJ, Wilson JM, Flotte TR. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci U S A 2009: 106(38): 16363-16368.
Stolk J, Tov N, Chapman KR, Fernandez P, MacNee W, Hopkinson NS, Piitulainen E, Seersholm N, Vogelmeier CF, Bals R, McElvaney G, Stockley RA. Efficacy and safety of inhaled alpha-1-antitrypsin in patients with severe alpha-1-antitrypsin deficiency and frequent exacerbations of Chronic Obstructive Pulmonary Disease. Eur Respir J 2019.




